This was my poster presentation from the 2023 American Thoracic Society International Conference in Washington D.C. from May 19 to 24.
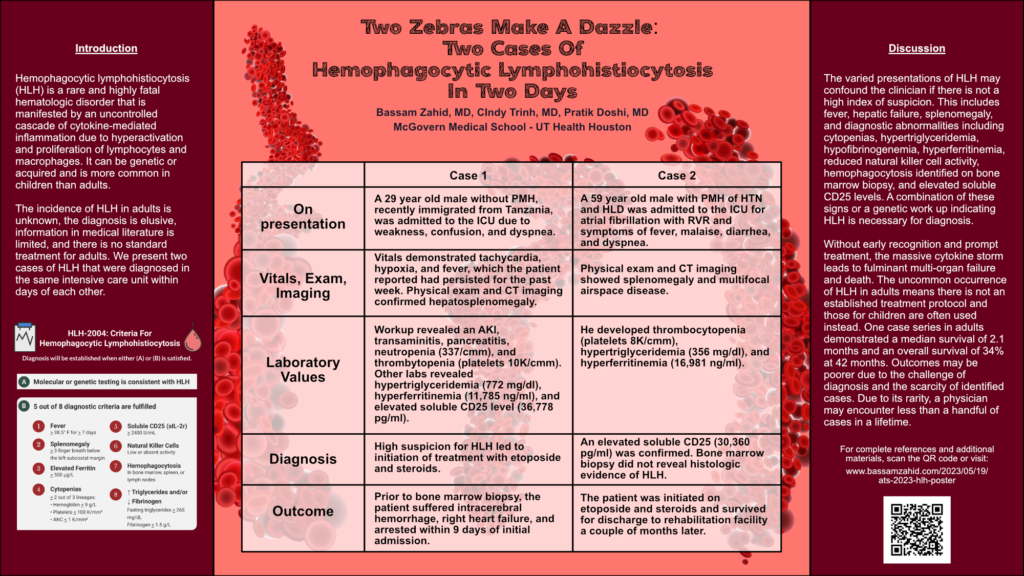
Introduction
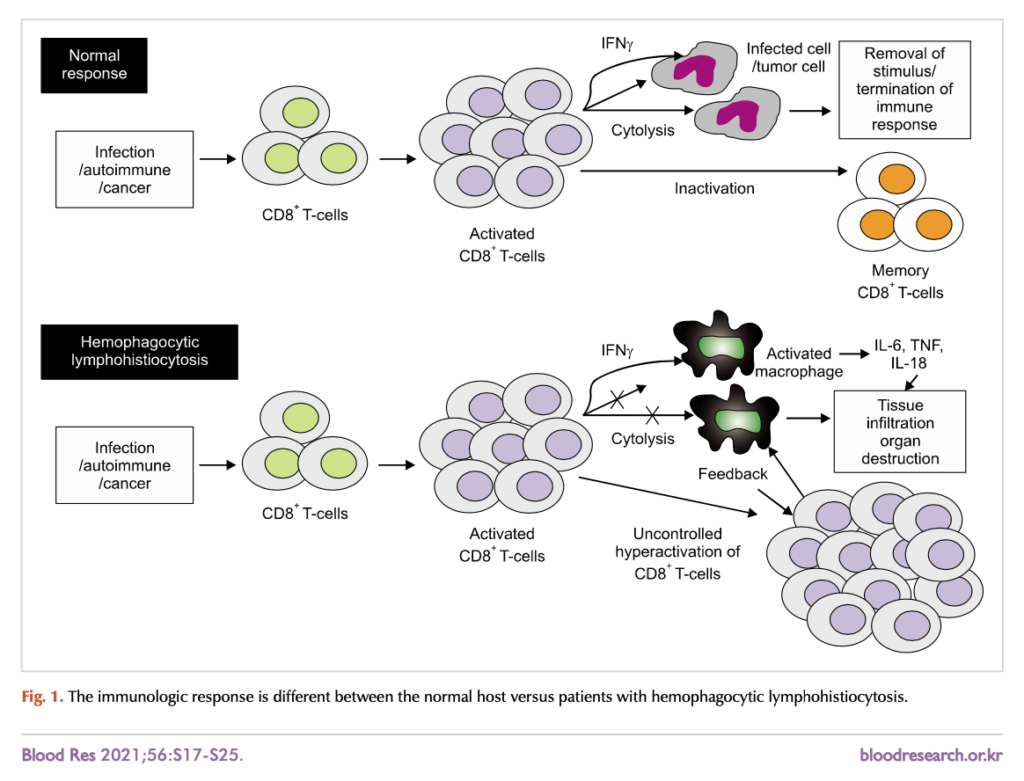
Hemophagocytic lymphohistiocytosis (HLH) is a rare and highly fatal hematologic disorder that is manifested by an uncontrolled cascade of cytokine-mediated inflammation due to hyperactivation and proliferation of lymphocytes and macrophages. It can be genetic or acquired and is more common in children than adults.
The incidence of HLH in adults is unknown, the diagnosis is elusive, information in medical literature is limited, and there is no standard treatment for adults. We present two cases of HLH that were diagnosed in the same intensive care unit within days of each other.
Case 1
- A 29 year old male without PMH, recently immigrated from Tanzania, was admitted to the ICU due to weakness, confusion, and dyspnea. Vitals demonstrated tachycardia, hypoxia, and fever, which patient reported had persisted for the past week. Physical exam and CT imaging confirmed hepatosplenomegaly.
- Further workup revealed an AKI, transaminitis, pancreatitis, neutropenia (337/cmm), thrombytopenia (platelets 10K/cmm). Other labs revealed hypertriglyceridemia (772 mg/dl), hyperferritinemia (11,785 ng/ml), and elevated soluble CD25 level (36,778 pg/ml).
- High concern for HLH led to initiation of treatment with etoposide and steroids.
- Prior to bone marrow biopsy, patient suffered intracerebral hemorrhage, right heart failure, and arrested within 9 days of initial admission.
Case 2
- A 59 year old male with PMH of HTN and HLD was admitted to the ICU for atrial fibrillation with RVR and symptoms of fever, malaise, diarrhea, and dyspnea.
- He subsequently developed thrombocytopenia (platelets 8K/cmm), hypertriglyceridemia (356 mg/dl), and hyperferritinemia (16,981 ng/ml).
- Physical exam and CT imaging showed splenomegaly and multifocal airspace disease.
- An elevated soluble CD25 (30,360 pg/ml) was confirmed. Bone marrow biopsy did not reveal histologic evidence of HLH.
- The patient was initiated on etoposide and steroids and survived for discharge to rehabilitation facility a couple of months later.
Discussion
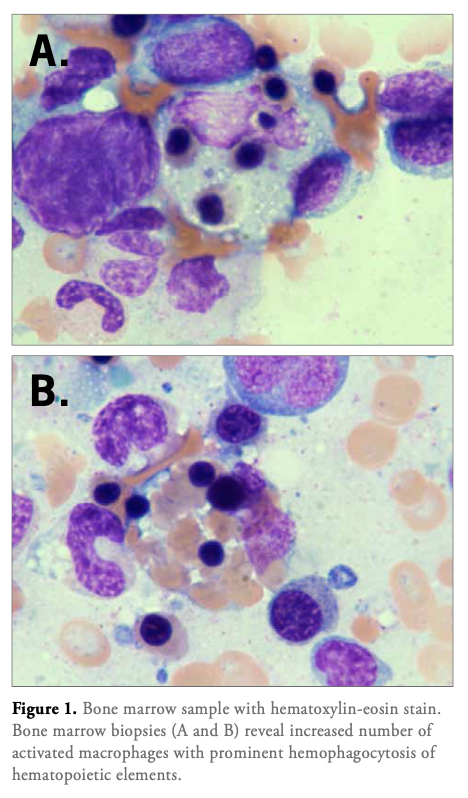
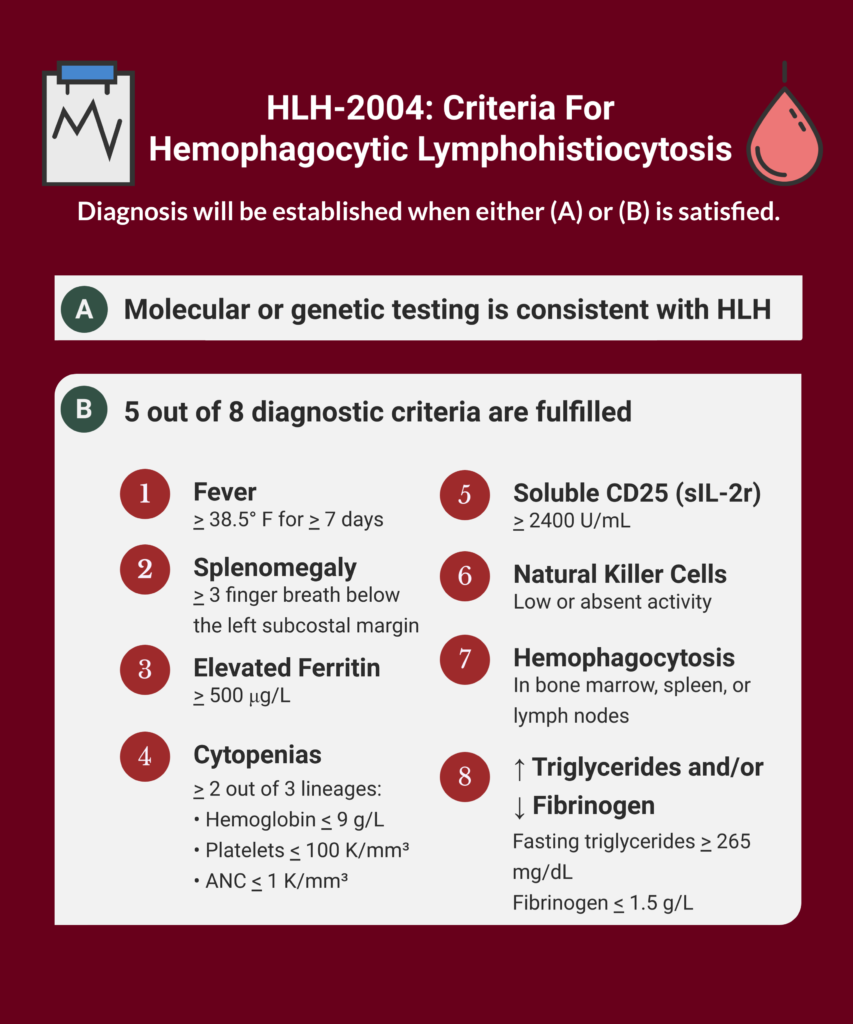
The varied presentations of HLH may confound the clinician if there is not a high index of suspicion. This includes fever, hepatic failure, splenomegaly, and diagnostic abnormalities including cytopenias, hypertriglyceridemia, hypofibrinogenemia, hyperferritinemia, reduced natural killer cell activity, hemophagocytosis identified on bone marrow biopsy, and elevated soluble CD25 levels. A combination of these signs or a genetic work up indicating HLH is necessary for diagnosis.
Without early recognition and prompt treatment, the massive cytokine storm leads to fulminant multi-organ failure and death. The uncommon occurrence of HLH in adults means there is not an established treatment protocol and those for children are often used instead. One case series in adults demonstrated a median survival of 2.1 months and an overall survival of 34% at 42 months. Outcomes may be poorer due to the challenge of diagnosis and the scarcity of identified cases. Due to its rarity, a physician may encounter less than a handful of cases in a lifetime.
References
- Henter, J.I., et al., HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer, 2007. 48(2): p. 124-31.
- Favara, B.E., Hemophagocytic lymphohistiocytosis: a hemophagocytic syndrome. Semin Diagn Pathol, 1992. 9(1): p. 63-74.
- Parikh, S.A., et al., Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc, 2014. 89(4): p. 484- 92.
- Kim, Y.R., Kim, D.Y. Current status of the diagnosis and treatment of hemophagocytic lymphohistiocytosis in adults. Blood Res 2021; 56(S1): S17-S25.
- Kleynberg, R.L., Schiller, G.J. Secondary hemophagocytic lymphohistiocytosis in adults: an update on diagnosis and therapy. Clinical Advances in Hematology & Oncology, Nov 2012. 10(11): p. 726-732.